Basile Paolo, 1 Napoli Gianluigi 1, Tricarico Giuseppe 1, Falco Giorgia 1, Carella Maria Cristina 1, Anaclerio Matteo 1, Forleo Cinzia 1, Guaricci Andrea Igoren1.
1 U.O. Cardiologia Universitaria, Dipartimento di Emergenza e dei Trapianti d’Organo (DETO), Università di Bari “Aldo Moro”, Piazza G. Cesare 11, Bari (BA), 70124, Italy
ABSTRACT
La mutazione R222Q nel gene SCN5A è stata recentemente correlata alla sindrome MEPPC (multifocal ectopic Purkinje-related premature contractions) in cui, tramite un meccanismo di attività triggerata nelle fibre di Purkinje, la mutazione gain-of-function nel canale del sodio voltaggio-dipendente Nav1.5 determina extrasistoli atriali e ventricolari polimorfe e ripetitive con possibile evoluzione in cardiopatia dilatativa e/o morte improvvisa.
Il presente case report descrive un cluster familiare di sindrome MEPPC ad esordio con elevato burden aritmico non responsivo né ai beta-bloccanti né all’ablazione transcatetere dell’extrasistolia ventricolare. Il trattamento con flecainide ha invece determinato l’abolizione completa del fenotipo aritmico.
INTRODUZIONE
La sindrome MEPPC (multifocal ectopic Purkinje-related premature contractions) è stata recentemente associata alla mutazione autosomica dominante R222Q-SCN5A che, mediante un meccanismo di gain-of-function del gene SCN5A, determina una ripolarizzazione incompleta nelle cellule di Purkinje triggerando potenziali d’azione prematuri che si propagano ai ventricoli1, 2 con un elevato tasso di extrasistoli ventricolari polimorfe e tachicardie ventricolari non sostenute (TVNS).
Descriviamo un cluster familiare di sindrome MEPPC esordita con un elevato burden aritmico in un paziente di 21 anni.
PRESENTAZIONE DEI CASI
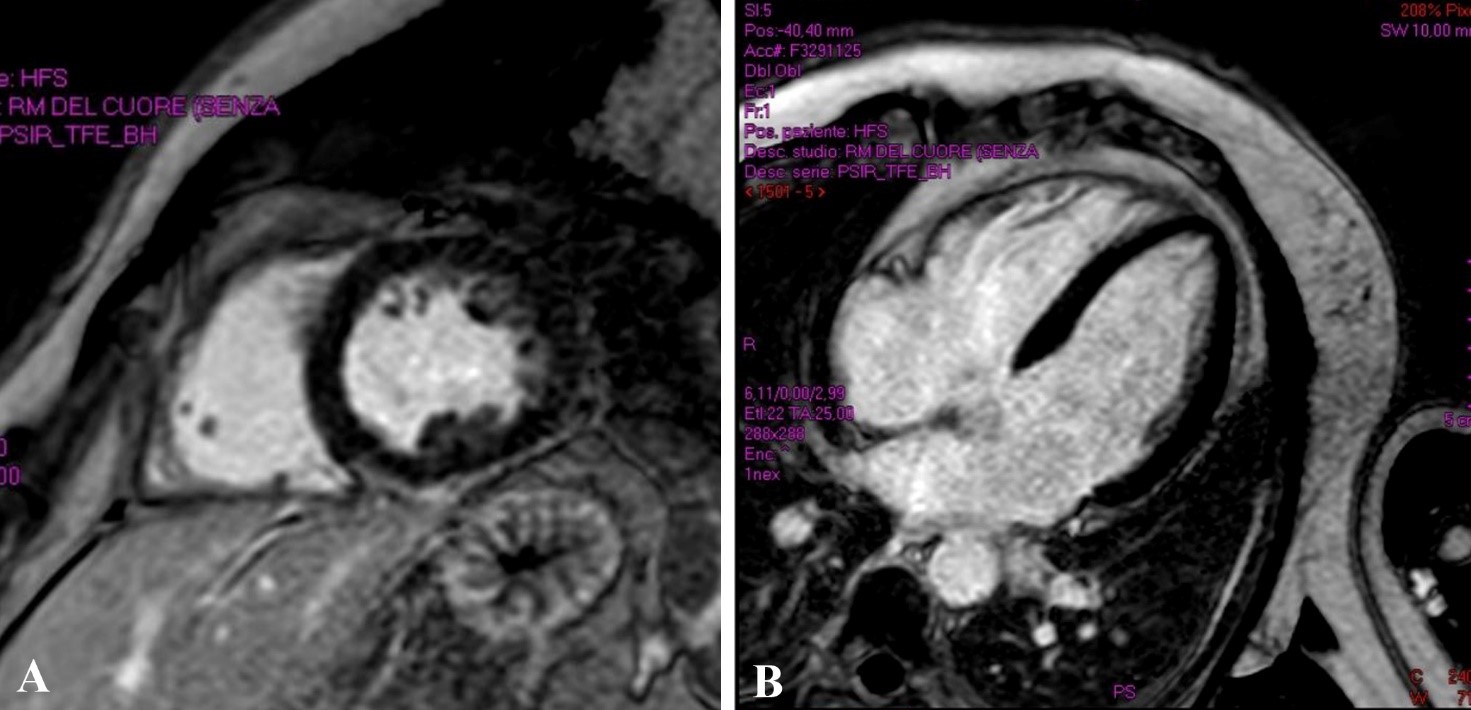
Un paziente maschio di 21 anni è stato indirizzato presso il nostro centro per episodi sincopali non traumatici, a riposo, preceduti da sintomatologia neurovegetativa, con successivo riscontro all’ECG-Holter di periodi di ritmo giunzionale, idioventricolare e di blocco atrio-ventricolare di II° grado tipo 1 associati a TVNS polimorfe per le quali era stato avviato, con risultati trascurabili, trattamento con nadololo e flecainide a dosaggio ridotto (50 mg x2/die). Il paziente ha riferito familiarità per cardiomiopatia dilatativa (CMD) e morte cardiaca improvvisa (MCI), negando abitudine tabagica o uso di droghe. L’esame obiettivo, le indagini di laboratorio e l’ECG di ingresso sono risultati nella norma. Né l’ecocardiogramma né la risonanza magnetica (RMN) cardiaca hanno evidenziato anomalie strutturali (Figura 1).
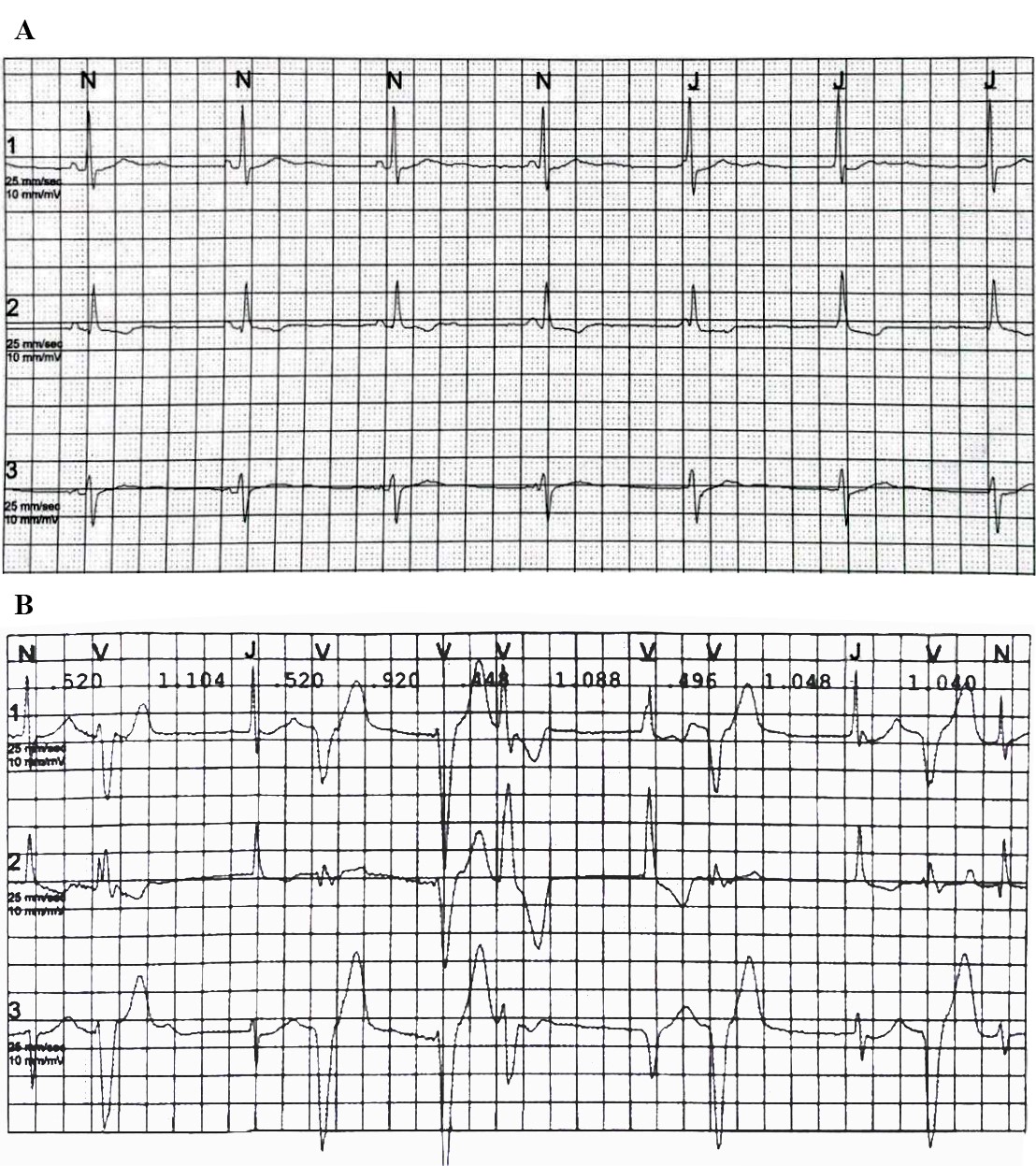
Figura 2. ECG dinamico sec Holter delle 24 ore del padre del paziente. A, Ritmo giunzionale con dissociazione atrioventricolare isoritmica. B, Tachicardia ventricolare polimorfa non sostenuta.
Si è pertanto deciso di incrementare la posologia della flecainide (100 mg x2/die) e di procedere all’impianto di un loop-recorder. Nel follow-up a 6 mesi si è assistito alla completa remissione degli episodi aritmici. L’analisi genetica condotta durante il ricovero ha successivamente rivelato la mutazione R222Q-SCN5A, con successivo avvio dello screening genetico familiare e riscontro positivo nel padre.
L’uomo, di 56 anni, affetto da ipertensione arteriosa e dislipidemia mista, ha negato abitudini voluttuarie, sintomatologia anginosa, dispnea, cardiopalmo e/o sincopi. La valutazione iniziale è risultata anche in questo caso nella norma. L’ECG-Holter ha evidenziato periodi di ritmo giunzionale con dissociazione atrioventricolare isoritmica e un elevato numero di extrasistoli ventricolari polimorfe e TVNS provenienti principalmente dal tratto di efflusso del ventricolo destro (Figura 2), regredite durante sforzo e scarsamente responsive al metoprololo.
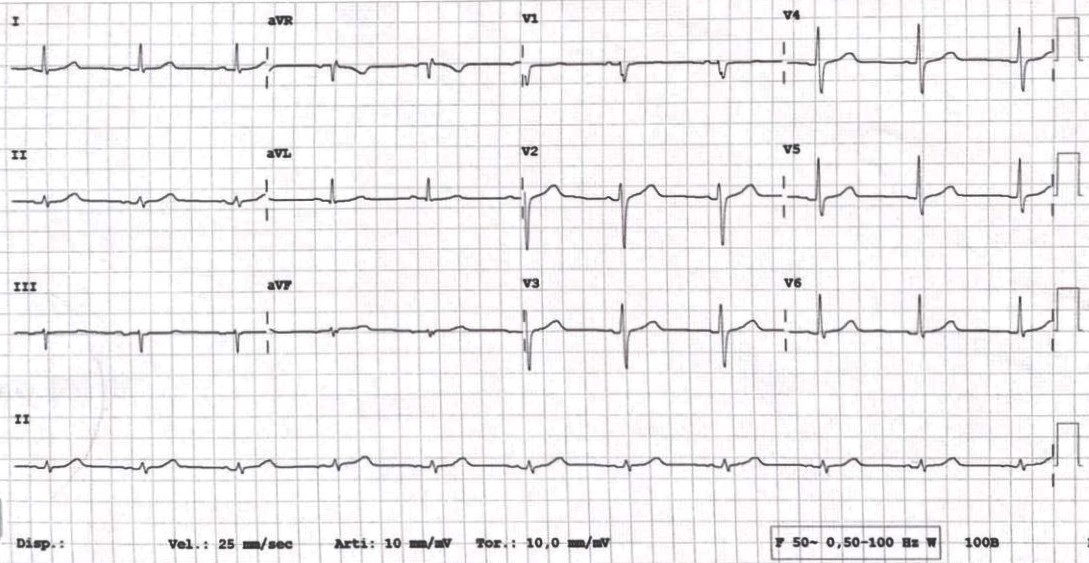
Per il riscontro ecocardiografico di lieve dilatazione e disfunzione sistolica del ventricolo sinistro, in assenza di fibrosi miocardica alla RMN cardiaca e di cause ischemiche alla coronarografia, si è optato per l’ablazione transcatetere dell’extrasistolia. Nonostante la regressione della disfunzione sistolica, l’efficacia dell’ablazione sul burden aritmico a 3 mesi è risultata trascurabile. Si è proceduto pertanto a testare la flecainide per via endovenosa (2 mg/Kg) con immediata regressione dell’extrasistolia ventricolare (Figura 3).
A 2 settimane dall’inizio della flecainide (100 mg x2/die), l’ECG-Holter ha confermato l’efficacia del farmaco (Tabella 1). Entrambi i pazienti sono attualmente in follow-up presso il nostro centro.
Tabella 1. ECG dinamico delle 24 ore del padre del paziente. Si può notare il netto decremento delle tachiaritmie sopraventricolari e ventricolari dopo assunzione della flecainide rispetto ai precedenti interventi terapeutici.
| Extrasistoli Atriali (n) | Extrasistoli Ventricolari (n) | TVNS (n) | |
| Senza terapia | 1315 | 7475 | 1 |
| Con beta-bloccante | 0 | 3776 | 1 |
| Post-ablazione transcatetere | 632 | 2496 | 127 |
| Con flecainide | 33 | 0 | 0 |
TVNS: tachicardia ventricolare non sostenuta.
DISCUSSIONE
Le mutazioni del gene SCN5A possono determinare un ampio spettro di sindromi: Brugada, QT lungo, CMD, e la rara sindrome MEPPC2-9. Quest’ultima è caratterizzata da una gain-of-function nel canale del sodio voltaggio-dipendente Nav1.510 che determina una ripolarizzazione incompleta nelle cellule di Purkinje generando potenziali d’azione prematuri che vengono condotti in via anterograda o retrograda a ventricoli o agli atri8 con un fenotipo clinico caratterizzato da extrasistoli ventricolari polimorfe ripetitive, tachiaritmie atriali, ritmo giunzionale, CMD potenzialmente reversibile e MCI. Non sono stati descritti né prolungamento dell’intervallo QT né sopraslivellamento del tratto ST2, 3, 5.
Il riscontro di tachiaritmie ventricolari necessita sempre una approfondita diagnosi differenziale. L’ECG da sforzo, eseguito nel padre, è un valido strumento diagnostico per escludere la tachicardia ventricolare polimorfa catecolaminergica. La coronaro-TC, la scintigrafia miocardica stress-rest o la coronarografia sono invece necessarie per escludere un’eziologia ischemica, specialmente nei pazienti con fattori di rischio cardiovascolare11. L’ablazione transcatetere è attualmente indicata nel sospetto di tachicardiomiopatia12, ma nel nostro caso non ha ottenuto gli effetti desiderati. Anche i beta-bloccanti, farmaci di prima linea nella gestione dell’extrasistolia, hanno sortito un modesto effetto terapeutico11. Dall’analisi della letteratura è emerso che i bloccanti dei canali del sodio come l’idrochinidina (classe IA) o la flecainide (classe IC), previa valutazione in regime ospedaliero della sicurezza e tollerabilità del farmaco, sono efficaci nel ridurre il burden aritmico e nel far regredire la disfunzione sistolica, quando presente2, 3, 5, 9. Il beneficio clinico sembrerebbe confermato anche al follow-up a lungo termine13. Attualmente non ci sono evidenze su un ruolo protettivo del defibrillatore impiantabile in questo contesto e ulteriori studi sono necessari per definire la gestione ottimale di questa rara sindrome.
CONCLUSIONE
La presenza di numerose extrasistoli ventricolari e TVNS polimorfe, in presenza di familiarità per CMD e MCI, in un cuore strutturalmente normale o con segni di lieve disfunzione sistolica e dilatazione del ventricolo sinistro, in assenza di coronaropatia o fibrosi miocardica, può sollevare il sospetto della sindrome MEPPC. In tal caso il riscontro al test genetico della mutazione gain-of-function R222Q-SCN5A del canale Nav1.5 offre la conferma diagnostica e può indirizzare verso una terapia esclusivamente medica con flecainide. I beta bloccanti sono di dubbia utilità in questo contesto e l’ablazione transcatetere ha scarsi benefici a causa dei molteplici focolai di origine delle extrasistoli.
BIBLIOGRAFIA
1. Elliott, P.M., Multifocal ectopic Purkinje-related premature contractions: Sorting the wheat from the chaff. International Journal of Cardiology, 2018. 257: p. 218-219.
2. Laurent, G., et al., Multifocal ectopic Purkinje-related premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol, 2012. 60(2): p. 144-56.
3. Doisne, N., et al., A novel gain-of-function mutation in SCN5A responsible for multifocal ectopic Purkinje-related premature contractions. Hum Mutat, 2020. 41(4): p. 850-859.
4. Leventopoulos, G., et al., You cannot ablate the Lernaean Hydra: SCN5A mutation in a patient with multifocal ectopic Purkinje-related premature contractions syndrome treated with Flecainide and an implant of a subcutaneous defibrillator-a case report. Eur Heart J Case Rep, 2021. 5(4): p. ytab158.
5. Mann, S.A., et al., R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol, 2012. 60(16): p. 1566-73.
6. McNair, W.P., et al., SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation, 2004. 110(15): p. 2163-7.
7. Ter Bekke, R.M.A., et al., Beauty and the beat: A complicated case of multifocal ectopic Purkinje-related premature contractions. HeartRhythm Case Rep, 2018. 4(9): p. 429-433.
8. Wilde, A.A.M. and A.S. Amin, Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin Electrophysiol, 2018. 4(5): p. 569-579.
9. Zakrzewska-Koperska, J., et al., Rapid and effective response of the R222Q SCN5A to quinidine treatment in a patient with Purkinje-related ventricular arrhythmia and familial dilated cardiomyopathy: a case report. BMC Med Genet, 2018. 19(1): p. 94.
10. Remme, C.A., Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol, 2013. 591(17): p. 4099-116.
11. Priori, S.G., et al., 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J, 2015. 36(41): p. 2793-2867.
12. Pedersen, C.T., et al., EHRA/HRS/APHRS expert consensus on ventricular arrhythmias. Europace, 2014. 16(9): p. 1257-83.
13. Peters, S., et al., Long-Term Efficacy and Safety of Sodium Channel Antagonists in Patients With p.R222Q <i>SCN5A</i>-Related Arrhythmic Dilated Cardiomyopathy. JACC: Clinical Electrophysiology, 2021. 7(1): p. 126-128.