Vincenzo Battaglia*, MD; Dario Donia*, MD; Alessia Chiara Latini*, MD; Gianluca Mincione*, MD; Cristina Panico, PhD; Elena Corrada, MD; Lorenzo Monti, MD; Antonio Frontera, PhD.
IRCCS Humanitas, Rozzano, Italia
Abstract
La cardiomiopatia aritmogena (ACM) può manifestarsi durante il follow-up con un quadro atipico (definito “hot phase”) caratterizzato da dolore toracico, rilascio di enzimi di miocardiocitonecrosi ed alterazioni elettrocardiografiche, in assenza di anomalie coronariche. Diverse evidenze supportano l’ipotesi che episodi ricorrenti simil-miocarditici possano accompagnare l’evoluzione della malattia in soggetti geneticamente predisposti1.
Il caso presentato è emblematico di come attraverso il processo infiammatorio siano stati indotti dei cambiamenti fenotipici, dirimenti nel porre diagnosi definitiva di ACM, sebbene inizialmente tale ipotesi diagnostica fosse poco sospetta.
Caso clinico
Il paziente è un ragazzo di 17 anni che accedeva in PS per arresto cardiocircolatorio (ACC) testimoniato, durante partita di pallacanestro, in assenza di prodromi. Al monitor veniva registrata fibrillazione ventricolare, interrotta da singola scarica elettrica (1 DC-shock) con successivo ripristino della circolazione spontanea (ROSC).
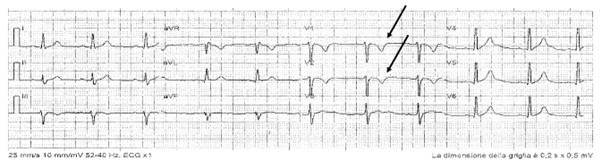
In anamnesi non compariva familiarità per malattie cardiovascolari o morte cardiaca improvvisa (SCD). Entrambi i genitori non presentavano precedenti cardiologici. L’anamnesi patologica remota era muta. All’ingresso, i parametri vitali e l’esame obiettivo risultavano nella norma. Si segnalava minimo movimento delle troponine, in progressiva riduzione nei giorni successivi al ricovero (da 26,1 ng/L a 8,2 ng/L), assenza di disionie, non compromissione della funzione renale. All’ECG d’ingresso si registrava ritmo sinusale con lieve ritardo di conduzione intraventricolare destro (terminal activation duration <55 ms, misurato dal nadir dell’onda S alla fine del QRS), con onde T invertite in V1-V2, in assenza di QRS frammentato o onde epsilon (figura 1).
All’ecocardiogramma transtoracico si documentava una lieve dilatazione del ventricolo destro (ATDi=14.7cm2/m2) con conservata funzione di pompa (TAPSE 21 mm), in assenza di chiare aree di bulging/aneurisma/discinesia. La.TC coronarica escludeva origine anomala delle coronarie. Alla RM cuore basale si evidenziava lieve dilatazione biventricolare (VTD VS = 210 ml, VTD VD = 270 ml) con funzione sistolica ai limiti inferiori (FE VS = 55 %, FE VD = 45%), in assenza di alterazioni miocardiche tissutali o della cinesi segmentaria. Tempo T1 (circa 1000±30 ms) e T2 (circa 45±4 ms) miocardico di valore globalmente normale, in assenza di aree di delayed enhancement.
Venivano eseguiti i test provocativi con flecainide (2 mg/kg somministrati in 10 minuti, con valutazione ripetitiva del tracciato con derivazioni standard e derivazioni precordiali ‘alte’) ed isoproterenolo (infusione di 45 µg/min in 3 minuti) che risultavano rispettivamente non conclusivi per pattern di Brugada e/o tachiaritmie ventricolari sostenute indotte. All’ ECG-Holter delle 24 ore, si documentava extrasistolia ventricolare (BEV isolati 1252, di cui almeno 500 a morfologia BBSn ad asse superiore).
Allo studio elettrofisiologico endocavitario, durante la stimolazione adrenergica con isoproterenolo, si osservava l’insorgenza di extrasistolia ventricolare di due diverse morfologie: la prima con morfologia compatibile con origine dal sistema di Purkinje destro (tipo BBSn, positività in D1, aVL. Negatività in DII, DIII, aVF. Piccola onda R in V1. Transizione in V6; la seconda morfologia (meno frequente) compatibile con origine dal tratto efflusso del ventricolo destro (RVOT) (transizione in V4, BBSn, asse inferiore).
A seguito di decisione collegiale, in prevenzione secondaria, veniva impiantato defibrillatore sottocutaneo (S-ICD).
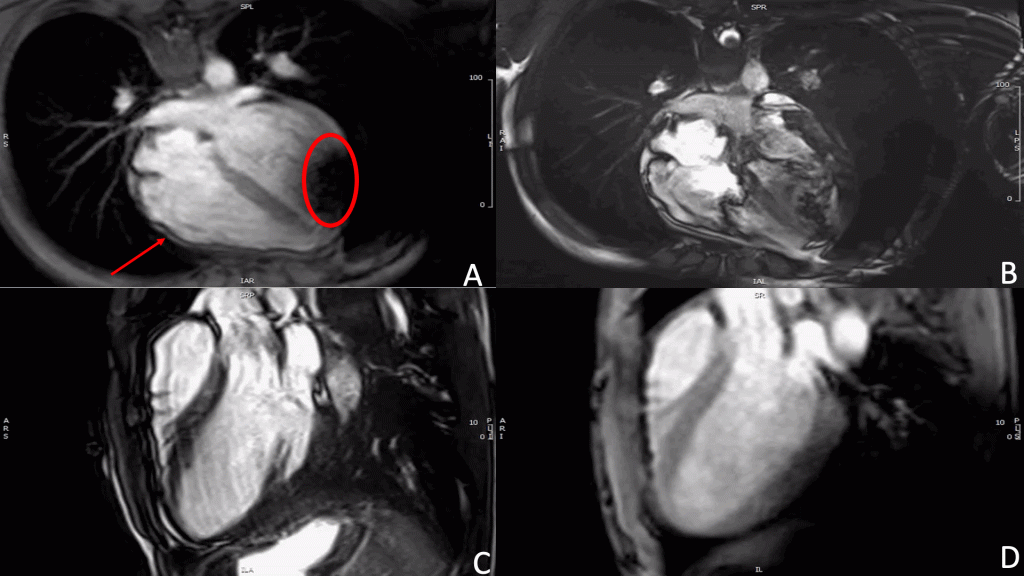
Dopo alcuni giorni di degenza, a distanza dalle procedure interventistiche, si riscontrava occasionalmente, in assenza di sintomi, incremento dei valori di troponina ad elevata sensibilità (hsTnI), con picco fino a 30.000 ng/L. All’ECG veniva registrata un’accentuata inversione dell’onda T in V2. Alla coronarografia eseguita successivamente si documentava albero coronarico esente da stenosi, test all’acetilcolina negativo per vasospasmo e ventricolografia destra con aspetto “a pila di piatti”8. Con i limiti procedurali legati all’impianto dell’S-ICD, si ripeteva RM che mostrava, rispetto alla precedente, una riduzione della frazione d’eiezione del ventricolo sinistro (FE VS 52%) con comparsa di ipocinesia della parete inferiore e infero-laterale nei segmenti medio-apicali, contestualmente ad una dilatazione del ventricolo destro con funzione di pompa depressa (FE VD 34%), in presenza di area acinetica in apice e di ipocinesia del tratto di afflusso (figura 2).
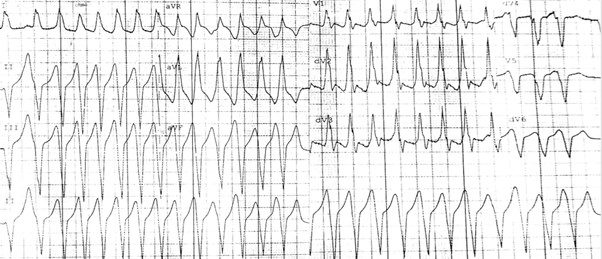
Al monitoraggio telemetrico, veniva segnalata la presenza di extrasistoli ventricolari, in un caso organizzate in breve run di tachicardia ventricolare non sostenuta (TVNS), ad asse superiore e morfologia a blocco di branca destra; al successivo ECG di superficie si aveva presenza di onde T negative da V4 a V6, reperti non riscontrati precedentemente.
Dunque, in accordo con i criteri di Padova 2020, si poneva diagnosi clinica definitiva per cardiomiopatia aritmogena (ACM) biventricolare7.
Discussione
La cardiomiopatia aritmogena è una malattia primitiva del miocardio che coinvolge il ventricolo destro, sinistro o entrambi, caratterizzata da sostituzione fibro-adiposa di tessuto miocardico, in grado di condizionare una disfunzione ventricolare globale e/o regionale e predisporre a potenziali aritmie ventricolari fatali, soprattutto nei giovani atleti, indipendentemente dalla severità della disfunzione. La prevalenza della patologia si stima essere 1:2000-1:5000 nella popolazione generale7. Studi di genetica molecolare hanno dimostrato un‘associazione tra l’ACM e alterazioni dei geni codificanti per le proteine strutturali delle giunzioni intercellulari: quelle più comuni sono a carico delle proteine desmosomiali, come placofillina (PKP2), desmoplachina (DSP), desmogleina (DSG2), e in misura minore delle proteine non desmosomiali, come fosfolambano (PLN), filamina-C (FLNC), laminina A/C (LMNA)1.
Sebbene la malattia nella sua forma “classica” (ARVC) si caratterizzi per un precoce coinvolgimento del ventricolo destro, con un interessamento del ventricolo sinistro nelle fasi avanzate, in letteratura ci sono crescenti evidenze di varianti fenotipiche con precoce coinvolgimento del ventricolo sinistro, che può evolvere parallelamente (variante “biventricolare”), come il caso riportato, o prevalere rispetto a quello del ventricolo destro (variante a “dominanza sinistra”, ALVC).
Data l’assenza di alterazioni patognomoniche per la malattia, in accordo con i criteri di Padova 2020, la diagnosi di ACM si basa su un approccio multiparametrico che tiene conto di parametri morfo-funzionali e strutturali, elettrocardiografici, di imaging e genetici, nonché della storia clinica e familiare del paziente, raggruppati in 6 categorie1,7.
Nel caso esaminato, il paziente all’ingresso non manifestava un fenotipo conclamato per forma “classica” di cardiomiopatia aritmogena, sia per la modalità atipica d’esordio (ACC secondario a fibrillazione ventricolare, più caratteristico di ALVC)8, sia per il soddisfacimento di un solo criterio minore di ARCV – presenza di onde T negative in V1 e V2 in assenza di blocco di branca destra completo.
Pertanto, l’approccio adottato è stato quello di condurre degli accertamenti diagnostici volti ad escludere le principali cause di aritmie ventricolari maligne nella popolazione giovanile.
L’assenza di ipertrofia settale interventricolare all’ecoscopia permetteva di escludere una cardiomiopatia ipertrofica, prima causa epidemiologica di SCD nel giovane atleta.3
Anche in considerazione del fatto che la risonanza magnetica cardiaca non evidenziasse inizialmente alterazioni della cinesi, presenza di late gadolinium enhancement (LGE) o franche alterazioni morfo-strutturali, il fenotipo non lasciava propendere per una specifica cardiomiopatia.
All’ECG di superficie l’intervallo QT corretto risultava nei limiti di norma, dunque non suggestivo di sindrome da QT lungo (LQTS) o da QT corto (SQTS), e non evidenziava sopraslivellamento del punto J, nel sospetto di early repolarization syndrome. 9
Il test alla flecainide non ha slatentizzato un pattern di Brugada. 10 Allo stesso modo, al fine di valutare la risposta al trigger adrenergico, potenzialmente responsabile dell’arresto nel sospetto di tachicardia ventricolare polimorfa catecolaminergica (CPVT), si decideva di sottoporre il paziente a test provocativo con isoproterenolo: non è stata indotta alcuna aritmia ventricolare sostenuta 11. Veniva unicamente osservata la comparsa di notched T-waves parallelamente ad incremento dell’intervallo QT corretto rispetto al basale (461 ms versus 425 ms): benchè questi ultimi reperti facessero propendere per un fenotipo LQTS2, il trigger psico-fisico – e non sensoriale (uditivo) – dell’evento aritmico poco correlava con tale sindrome.
Escluse cause cardiache strutturali, ischemiche e primitive, in assenza di un substrato di FV, si prendeva in considerazione la fibrillazione ventricolare idiopatica (IVF) come plausibile eziologia (causa di SCD nei giovani adulti dal 14% al 23% dei casi)12: tale ipotesi era avvalorata dal fatto che alla telemetria e al SEF fossero stati registrati BEV isolati, con verosimile origine dalle fibre destre del Purkinje e da RVOT, tra i siti più frequentemente coinvolti nella genesi di IVF.4
Solamente in seguito al sostanziale rialzo troponinico in assenza di dolore toracico, alla luce delle alterazioni regionali della cinesi e delle dilatazioni di entrambi i ventricoli – riscontrate alla successiva RM – e dei concomitanti eventi aritmici a partenza dai segmenti del ventricolo sinistro morfostrutturalmente alterati, è stato possibile porre diagnosi “definitiva” di ACM nella variante biventricolare.
Analogamente al caso presentato, è stata recentemente descritta in letteratura una forma simil-miocarditica – piuttosto atipica – di presentazione della malattia (“hot phase”)1,6, frequentemente segnalata nei giovani (27±16 anni).
CONCLUSIONI
Nell’ambito dello spettro fenotipico della ACM, i dati strumentali ed elettrocardiografici inizialmente raccolti apparivano maggiormente suggestivi per un variante destra (ARVC): ciononostante, come descritto in letteratura la modalità d’esordio dell’ACC (FV) poco correlava con una forma classica della patologia7. Il caso riportato enfatizza come solo esclusivamente a seguito di un evento simil-miocarditico, proprio della storia naturale della malattia (“hot phase”), sia stato slatentizzato un fenotipo prima misconosciuto con coinvolgimento del ventricolo sinistro, giustificando la fibrillazione ventricolare come onset clinico.
Bibliografia
- ‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy – PubMed (nih.gov) 2021 Jun 7;23(6):907-917. doi: 10.1093/europace/euaa343.
- Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers https://academic.oup.com/eurheartj/article/36/14/847/540212?login=true
- 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society https://doi.org/10.1016/j.jacc.2017.10.054
- Mapping and Ablation of Idiopathic Ventricular Fibrillation – PubMed (nih.gov) PMID (PMC6153961) doi: 10.3389/fcvm.2018.00123
- Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review Circulation (ahajournals.org) https://doi.org/10.1161/CIRCULATIONAHA.121.055890
- An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis – PubMed (nih.gov) doi: 10.1093/eurheartj/ehy567
- Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria – PubMed (nih.gov) doi: 10.1016/j.ijcard.2020.06.005
- MR and CT imaging of Arrhythmogenic Cardiomyopathy Card Electrophysiol Clin. 2011 June 1; 3(2): 269–280. doi:10.1016/j.ccep.2011.02.002.
- Long QT Syndrome: A Comprehensive Review of the Literature and Current Evidence 2019 Mar;44(3)92-106. doi: 10.1016/j.cpcardiol.2018.04.002.
- Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome doi:10.1016/j.hrthm.2004.11.025
- CPVT: Arrhythmogenesis, Therapeutic Management, and Future Perspectives. A Brief Review of the Literature doi: 10.3389/fcvm.2019.00092
- Idiopathic Ventricular Fibrillation: Role of Purkinje System and Microstructural Myocardial Abnormalities 2020 Jun;6(6):591-608. doi: 10.1016/j.jacep.2020.03.010.