Light-chain cardiac amyloidosis: a case report of extraordinary sustained pathological response to cyclophosphamide, bortezomib, and dexamethasone combined therapy
Aldostefano Porcari 1*, Linda Pagura 1, Maddalena Rossi 1, Marika Porrazzo 2, Franca Dore 3, Rossana Bussani 4, Marco Merlo 1, and Gianfranco Sinagra 1
1 Center for Diagnosis and Treatment of Cardiomyopathies, Cardiovascular Department, Azienda Sanitaria Universitaria Giuliano-Isontina (ASUGI), University of Trieste, Via P. Valdoni 7, 34100, Trieste, Italy; 2 Department of Hematology, Azienda Sanitaria Universitaria Giuliano-Isontina (ASUGI), Trieste, Italy; 3 Department of Nuclear Medicine, Azienda Sanitaria Universitaria Giuliano-Isontina (ASUGI), University of Trieste, Trieste, Italy; and 4 Center for Diagnosis and Treatment of Cardiomyopathies, Cardiothoracic Department, Institute of Pathological Anatomy and Histology, Azienda Sanitaria Universitaria Giuliano-Isontina (ASUGI), University of Trieste, Trieste, Italy.
Abstract
Il coinvolgimento cardiaco è un fattore prognostico infausto nell’amiloidosi AL. Spesso preclude terapie curative, quali chemioterapia ad alte dosi e trapianto autologo di cellule staminali (ASCT). Il trapianto cardiaco può venire considerato prima dell’ASCT in casi selezionati. Nei pazienti non eleggibili per ASCT, la chemioterapia con ciclofosfamide, bortezomib e desametasone (CyBorD), anche a basso dosaggio, può portare a remissione dalla malattia con risposta cardiaca eccellente. Riportiamo il caso di una paziente di 50 anni, con forma avanzata di amiloidosi cardiaca AL, ritirata dalle liste trapianto (sia cardiaco che ASCT) in seguito a terapia CyBorD per eccezionale risposta ematologica e cardiaca persistente.
Introduzione
L’amiloidosi a catene leggere (AL) è la forma più comune di amiloidosi sistemica, caratterizzata dalla deposizione extracellulare di immunoglobuline monoclonali a catena leggera come proteine beta-fibrillari insolubili in vari tessuti, con conseguente insufficienza d’organo progressiva1. Il coinvolgimento cardiaco è comune (>50% dei pazienti con diagnosi di AL)2, e rappresenta il fattore prognostico peggiore. I pazienti con amiloidosi cardiaca avanzata (CA) di solito non traggono beneficio dai tradizionali trattamenti per l’insufficienza cardiaca3,4 e, spesso, non possono accedere a valide terapie curative come la chemioterapia ad alte dosi seguita dal trapianto autologo di cellule staminali (ASCT). La chemioterapia combinata con bortezomib è efficace nell’ottenere una risposta ematologica e d’organo in pazienti non idonei all’ASCT5, anche se la sopravvivenza globale nella CA avanzata è ancora bassa6. In casi selezionati, il trapianto di cuore (HTx) seguito da ASCT potrebbe rappresentare l’approccio adatto per ottenere una sopravvivenza a lungo termine e, talvolta, il pieno recupero dalla malattia ematologica7,8. Pertanto, è essenziale un’accurata selezione dei pazienti e una stratificazione del rischio.
Caso Clinico
La paziente è una donna di 50 anni con una storia recente di astenia, dispnea da sforzo e aumento di peso, con segni di grave insufficienza cardiaca di nuova insorgenza. Alla presentazione è ipotesa, con un soffio olosistolico meglio udibile all’apice cardiaco, rantoli polmonari bilaterali, edema degli arti inferiori e macroglossia. In anamnesi patologica remota risulta solo un intervento chirurgico al tunnel carpale bilaterale.
La diagnosi differenziale si pone con le cardiomiopatie, le cardiopatie ipertensive, ischemiche e valvolari. La macroglossia e la sindrome del tunnel carpale suggeriscono una malattia infiltrativa3, mentre il soffio cardiaco avalla la possibilità che una grave malattia valvolare possa precipitare l’insufficienza cardiaca acuta. L’assenza di fattori di rischio cardiovascolare rende improbabile la diagnosi di cardiopatia ischemica.
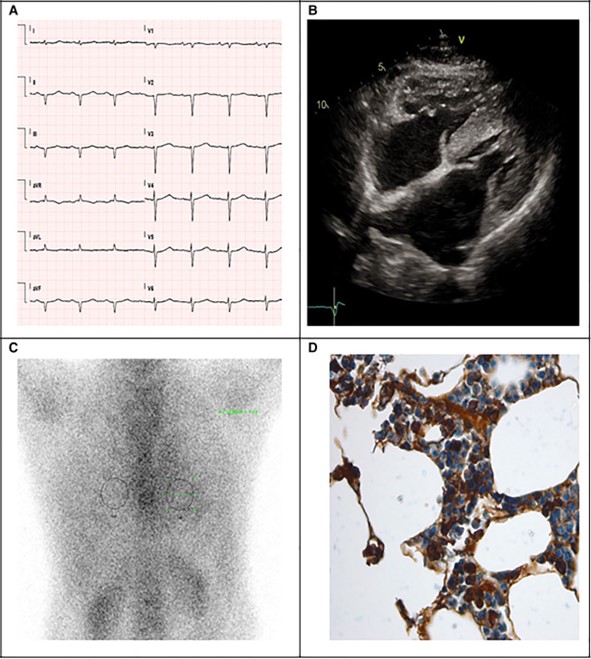
L’ECG mostra un ritmo sinusale con normali voltaggi e onde Q anteriori e inferiori (Figura 1A) non coerenti con il grado di ipertrofia del ventricolo sinistro (LV) (max 17 mm); all’ecocardiografia, si evidenziano normale cinetica (Figura 1B), disfunzione diastolica di grado III, GLS -14%, un pattern di “apical sparing”, lembi ispessiti della valvola mitrale, ipertrofia del ventricolo destro e vena cava inferiore dilatata.
Agli esami di laboratorio emergono BNP elevato (618 pg/L, v.n.100 pg/L) e una lieve troponinosi T (0,34 ng/mL, v.n. 0,25 ng/mL). La coronarografia esclude malattia coronarica.
Il sospetto di CA è elevato; viene seguito l’algoritmo di Gillmore9: le catene leggere libere λ sieriche (FLC) sono aumentate (108 mg/dL, delta FLC 93 mg/dL) e alla scintigrafia con difosfonato emerge un l’uptake miocardico Perugini grado 1 (Figura 1C). Si conferma istologicamente, con biopsia del grasso addominale, la diagnosi di AL-CA. Emerge una diagnosi concomitante di smoldering mieloma micromolecolare di tipo λ, sulla base del 22% di plasmacellule clonali alla biopsia del midollo osseo, senza segni di danno d’organo avanzato (Figura 1D).
Il coinvolgimento cardiaco avanzato (stadio cardiaco Mayo III)10 e il mieloma micromolecolare controindicano l’ASCT come terapia iniziale.
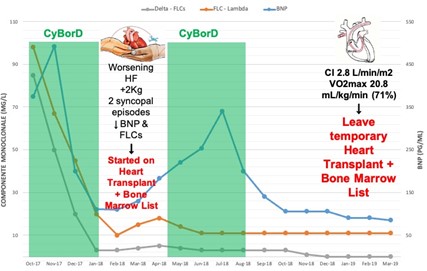
Pertanto, è stata avviata terapia citoriduttiva a basse dosi con ciclofosfamide, bortezomib e desametasone (CyBorD). Nel decorso, vi sono state frequenti riammissioni per scompenso cardiaco con deterioramento emodinamico. Sono stati rilevati un indice cardiaco di 2,5l/min/m2 e un consumo di ossigeno gravemente ridotto durante l’esercizio (VO2) picco (15,4 ml/kg/min, 50% valore previsto) con ridotta efficienza ventilatoria al test da sforzo cardiopolmonare.
Dopo un ciclo CyBorD, le strategie terapeutiche sono state discusse collegialmente, considerando tre variabili principali: giovane età, grave coinvolgimento cardiaco con prognosi infausta e assenza di significativo coinvolgimento extracardiaco, sistematicamente studiato. La paziente è stata giudicata un buon candidato per HTx seguito da ASCT. Pertanto, entro due mesi dalla diagnosi, è entrata in lista d’attesa per HTx. Ha avuto due episodi sincopali senza chiari prodromi, per cui è stato impiantato un defibrillatore in prevenzione primaria come ponte a HTx.
Discussione
Abbiamo riportato un caso insolito di AL-CA che ha lasciato le liste trapianto (sia cardiaca che ASCT) per risposta ematologica completa persistente (>3 anni) ed eccezionale miglioramento cardiaco in chemioterapia CyBorD. Da questo caso emergono i seguenti elementi:
(1) Mantenere un alto sospetto di CA nei pazienti con SC con FE conservata e red flags specifiche è essenziale per raggiungere una diagnosi precoce e iniziare terapie con impatto sulla sopravvivenza.
(2) La selezione della migliore strategia terapeutica si basa su un’accurata stratificazione prognostica multidisciplinare e quantificazione del carico di malattia cardiaca ed extracardiaca, che massimizza le possibilità di sopravvivenza.
(3) Il trattamento per LA deve essere personalizzato per il paziente.
Il sospetto clinico di CA sorge dall’integrazione di dati clinici, ecocardiografici e laboratoristici. In questo caso, la discrepanza tra i voltaggi del QRS all’ECG e il grado di ipertrofia del LV all’ecocardiografia, la severa disfunzione diastolica con il pattern “apical sparing”, la troponina persistentemente elevata e la storia di intervento al tunnel carpale bilaterale sono stati indizi di rilievo per la diagnosi. 2,11–13
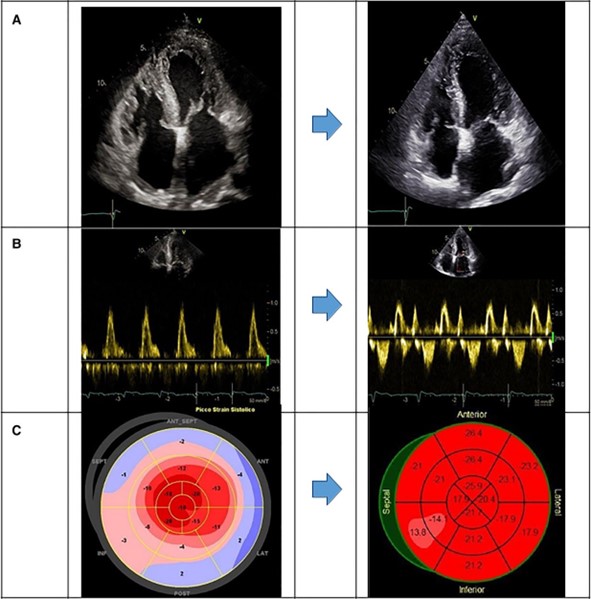
La stratificazione prognostica dei pazienti con AL-CA è stimata da punteggi dedicati che integrano i valori di NT-proBNP, troponina e FLC10 che possono essere utilizzati per monitorare la risposta al trattamento14. Alla nostra paziente è stato assegnato un punteggio Mayo stadio III, che delinea un coinvolgimento cardiaco avanzato con alto tasso di mortalità precoce. Spesso i pazienti con queste caratteristiche non hanno abbastanza tempo per rispondere alla terapia.
Nella fase acuta dell’amiloidosi AL, danno cardiaco e disfunzione derivano dalla deposizione miocardica di fibrille amiloidi e dall’effetto tossico delle catene leggere sui cardiomiociti, elementi reversibili con un’efficace chemioterapia. Pertanto, la sopravvivenza globale nell’amiloidosi AL dipende della risposta ematologica. La chemioterapia è diretta verso il clone sottostante e mira a sopprimere la produzione di FLC che causano disfunzione d’organo2. Attualmente, CyBorD è lo standard di cura per l’amiloidosi AL, e fornisce una risposta ematologica parziale (riduzione ≥50% delle concentrazioni di FLC) e una risposta cardiaca (riduzione >30% o >300 pg/mL di NT-proBNP) rispettivamente nel 60% e nel 25% circa dei pazienti trattati6,15. La sopravvivenza globale a 5 anni è dell’80%, con una durata mediana della risposta di 4,5 anni per i pazienti che hanno raggiunto una risposta dopo trattamento con solo CyBorD, in modo simile ai pazienti trattati in sequenza con CyBorD e ACST5. Comunemente, il regime CyBorD è difficilmente tollerato anche a basse dosi in presenza di ipotensione e SC gravemente sintomatico.
Nel caso presentato, la paziente non era idonea all’ASCT a causa di una grave infiltrazione amiloide cardiaca ed è stata inserita in lista HTx alla luce dell’assenza di un coinvolgimento extracardiaco significativo, dell’assenza di altre comorbidità e della giovane età. Sebbene l’HTx consenta l’ASCT e possa conferire un vantaggio in termini di sopravvivenza7,8, comporta un tasso non trascurabile di complicanze, inclusa la morte, e può essere eseguito solo in pochi centri di riferimento. Da segnalare che il corretto timing per ASCT dopo HTx è tutt’ora oggetto di studio. In una recente serie di trapianti, alcuni pazienti non hanno avuto il tempo di ricevere ASCT dopo HTx a causa della progressione della malattia ematologica16; pertanto, questo trattamento è considerabile solo in pazienti ben selezionati senza amiloide extracardiaca significativa in cui si prevede che l’ASCT conferisca un beneficio sostanziale in termini di sopravvivenza, con un rischio relativamente basso di mortalità correlata al trattamento17.
Inaspettatamente, nella nostra paziente il regime CyBorD a basse dosi è risultato efficace nell’ottenere una risposta ematologica completa e un’impressionante risposta cardiaca, con un buono stato clinico a lungo termine. Tuttavia, nella gestione della nostra paziente sono stati essenziali rivalutazioni cliniche frequenti, monitoraggio rigoroso della risposta ai trattamenti e aggiustamenti terapeutici.
La combinazione di daratumumab e bortezomib si è recentemente dimostrata efficace nel trattamento dell’amiloidosi AL, rendendo promettente una futura ridefinizione dello standard di cura nell’amiloidosi AL 18,19. Sono necessarie ulteriori ricerche, considerando che i progressi nella farmacoterapia dell’amiloidosi AL avranno un grande impatto sulla gestione clinica dei pazienti, riducendo possibilmente la necessità di ASCT.
Conclusioni
La diagnosi precoce del coinvolgimento cardiaco in AL fornisce il più ampio accesso alle opzioni terapeutiche, migliorando la sopravvivenza. L’HTx può essere considerato prima dell’ASCT in casi rigorosamente selezionati quando l’AL-CA avanzato preclude l’idoneità all’ASCT. È interessante notare che nei pazienti non idonei all’ASCT, la chemioterapia con regime CyBorD, anche a basse dosi, può portare a una remissione duratura della malattia con un’eccellente risposta cardiaca.
Bibliografia
1. Porcari A, Falco L, Lio V, et al. Cardiac amyloidosis: do not forget to look for it. European Heart Journal Supplements 2020;22(Supplement_E):E142–7.
2. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis. J Am Coll Cardiol 2016;68(12):1323–41.
3. Porcari A, Merlo M, Rapezzi C, Sinagra G. Transthyretin amyloid cardiomyopathy: An uncharted territory awaiting discovery. European Journal of Internal Medicine 2020;82:7–15.
4. Tini G, Cappelli F, Biagini E, et al. Current patterns of beta‐blocker prescription in cardiac amyloidosis: an Italian nationwide survey. ESC Heart Failure 2021;8(4):3369–74.
5. Basset M, Milani P, Nuvolone M, et al. Sequential response-driven bortezomib-based therapy followed by autologous stem cell transplant in AL amyloidosis. Blood Advances 2020;4(17):4175–9.
6. Palladini G, Sachchithanantham S, Milani P, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood 2015;126(5):612–5.
7. Gillmore JD, Goodman HJ, Lachmann HJ, et al. Sequential heart and autologous stem cell transplantation for systemic AL amyloidosis. Blood 2006;107(3):1227–9.
8. Barrett CD, Alexander KM, Zhao H, et al. Outcomes in Patients With Cardiac Amyloidosis Undergoing Heart Transplantation. JACC: Heart Failure 2020;8(6):461–8.
9. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016;133(24):2404–12.
10. Kumar S, Dispenzieri A, Lacy MQ, et al. Revised Prognostic Staging System for Light Chain Amyloidosis Incorporating Cardiac Biomarkers and Serum Free Light Chain Measurements. Journal of Clinical Oncology 2012;30(9):989–95.
11. Witteles RM, Bokhari S, Damy T, et al. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC: Heart Failure 2019;7(8):709–16.
12. Porcari A, Pagura L, Longo F, et al. Prognostic significance of unexplained left ventricular hypertrophy in patients undergoing carpal tunnel surgery. ESC Heart Failure 2022;9(1):751–60.
13. Merlo M, Porcari A, Pagura L, et al. A national survey on prevalence of possible echocardiographic red flags of amyloid cardiomyopathy in consecutive patients undergoing routine echocardiography: study design and patients characterization — the first insight from the AC-TIVE Study. European Journal of Preventive Cardiology 2022;29(5):e173–7.
14. Pregenzer-Wenzler A, Abraham J, Barrell K, Kovacsovics T, Nativi-Nicolau J. Utility of Biomarkers in Cardiac Amyloidosis. JACC: Heart Failure 2020;8(9):701–11.
15. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood 2020;136(23):2620–7.
16. di Nora C, Sponga S, Ferrara V, et al. Emerging therapy in light-chain and acquired transthyretin-related amyloidosis: an Italian single-centre experience in heart transplantation. Journal of Cardiovascular Medicine 2021;22(4):261–7.
17. di Nora C, Livi U. Heart transplantation in cardiac storage diseases: data on Fabry disease and cardiac amyloidosis. Current Opinion in Organ Transplantation 2020;25(3):211–7.
18. Kastritis E, Palladini G, Minnema MC, et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. New England Journal of Medicine 2021;385(1):46–58.
19. Mikhael JR, Schuster SR, Jimenez-Zepeda VH, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood 2012;119(19):4391–4.
Enrico Baldi administrator
Info sull'autore