Andrea Faggiano 1, Marco Vicenzi 1, Stefano Carugo 1
1 Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico di Milano, Dipartimento di Medicina Interna, Unità Operativa di Cardiologia, Università di Milano, Italia.
Abstract
Presentiamo il caso clinico di una paziente di 70 anni con anamnesi di Sclerosi Sistemica a coinvolgimento multiorgano giunta alla nostra attenzione per effettuare un test da sforzo cardiopolmonare (CPET) per dispnea ingravescente. Il CPET mostrava una severa riduzione della capacità aerobica secondaria a spiccata limitazione cardiogena. L’ecocardiogramma evidenziava una severa ipertrofia ventricolare sinistra, asimmetrica, a prevalenza settale. Dall’anamnesi emergeva la familiarità per morte improvvisa, sindrome del tunnel carpale bilaterale, rottura non traumatica del capo-lungo del bicipite configurando così la difficile diagnosi differenziale fra tre entità cliniche: cardiopatia sclerodermica a fenotipo ipertrofico, sclerosi sistemica associata ad amiloidosi cardiaca, sclerosi sistemica associata a cardiomiopatia ipertrofica. L’approccio multimodale di imaging sequenziale, combinato all’esecuzione dell’analisi genetica ci permetteva di raggiungere la diagnosi di cardiomiopatia ipertrofica e di condurre lo screening genetico e fenotipico nei famigliari di primo grado della paziente.
Caso Clinico
La paziente del nostro caso clinico è una donna di 70 anni affetta dal 1988 di Sclerosi Sistemica (SS) con coinvolgimento multiorgano (fenomeno di Raynaud, esofagopatia, coinvolgimento intestinale, artrite, calcinosi, teleangectasie). In anamnesi mai segnalato coinvolgimento sclerodermico cardiaco e/o ipertensione polmonare. Giungeva alla nostra attenzione tramite il centro unico di prenotazione regionale per l’esecuzione di un test ergometrico cardiopolmonare (CPET), richiesto dal centro di riferimento in considerazione della dispnea ingravescente per sforzi lievi (classe NYHA II-III) lamentata dalla paziente negli ultimi mesi.
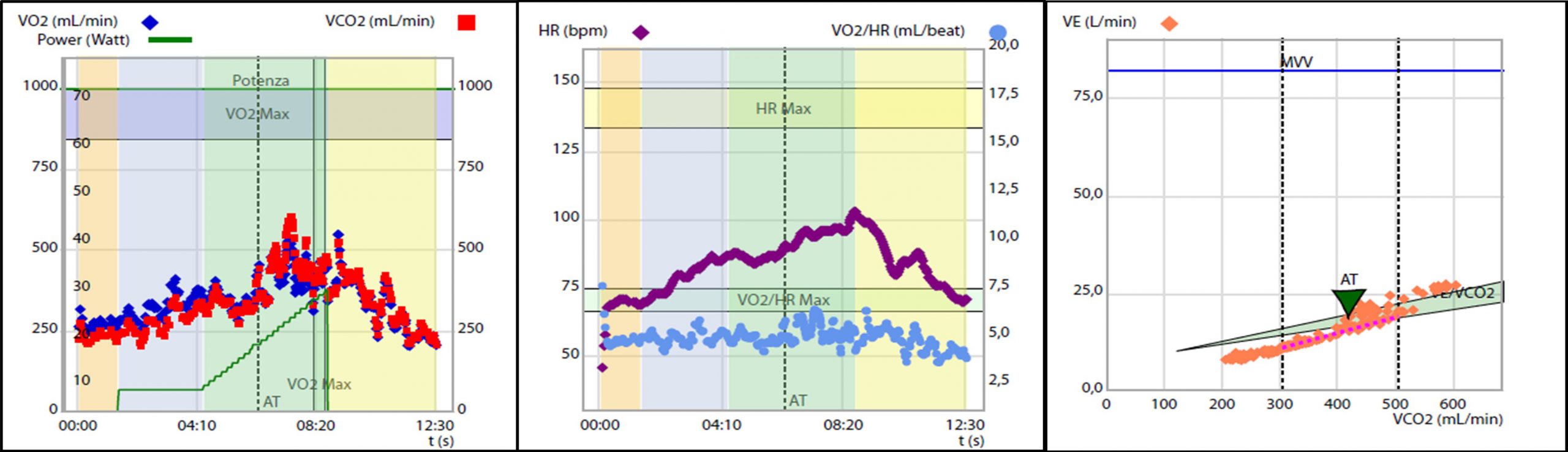
1A) Severa riduzione della capacità di esercizio aerobico certificata dal ridotto consumo di ossigeno al picco, pari a 9.9 ml/kg/min, ossia il 42% del predetto.
2A) Evidenza di severa limitazione cardiogena allo sforzo. Polso dell’ossigeno ridotto con plateau precoce e sostenuto, pari a 4 ml/battito, ossia il 60% del predetto.
3A) Inefficienza ventilatoria durante lo forzo, con slope del rapporto ventilazione -volume di anidride carbonica, VE/VCO2 slope pari a 39.9 ossia il 139% del predetto
Portava in visione l’ultimo ecocardiogramma, eseguito nel 2018, il quale mostrava lieve ipertrofia settale, normali dimensioni biventricolari, normale cinesi segmentaria e funzione sistolica globale, dilatazione atriale sinistra lieve, disfunzione diastolica di I° grado ed un basso rischio ecocardiografico di ipertensione polmonare. Le prove di funzionalità respiratorie recenti (2021) risultavano nella norma. L’elettrocardiogramma (ECG) basale effettuato prima del CPET rilevava segni di ipertrofia ventricolare sinistra con T negative in V3-V4-V5. Essendo la paziente di esile corporatura e tendente alla sarcopenia, il CPET veniva effettuato con una rampa blanda di incremento pari a 5 W/min.
L’esame veniva interrotto precocemente a 27 W per dispnea (BORG scale semplificata = 9) ed esaurimento muscolare (BORG scale semplificata = 8), risultava appena massimale per quoziente respirato (RQ = 1.1) e sottomassimale per frequenza cardiaca (FC = 103 bpm, 70% della massima frequenza cardiaca predetta). Dal CPET emergeva un quadro di severa riduzione della capacità di esercizio aerobico (consumo di ossigeno al picco, VO2 peak pari a 9.9 ml/kg/min, ossia il 42% del predetto. Figura 1 A) secondario a spiccata limitazione cardiogena (polso dell’ossigeno = 4 ml/battito, ossia il60% del predetto, con plateau precoce e sostenuto. Figura 1 B) associata ad inefficienza ventilatoria (slope del rapporto ventilazione – volume di anidride carbonica, VE/VCO2 slope = 39.9, ossia il 139% del predetto. Figura 1 C) in assenza di limitazione respiratoria.
In considerazione del quadro elettrocardiografico basale e del CPET si decideva di eseguire durante la stessa seduta un esame ecocardiografico (Panel 2) che mostrava una marcata ipertrofia asimmetrica del ventricolo sinistro, maggiore a carico del setto interventricolare (18 mm), in assenza di “systolic anterior movement” e di ostruzione dinamica all’efflusso, né in basale, né durante manovra di Valsalva ed in presenza di iniziale incremento del gradiente atrioventricolare destro (Tricuspid regurgitant Jet = 3 m/s).
Dati i reperti di imaging veniva ripercorsa l’anamnesi da cui emergeva la familiarità per morte improvvisa (sorella del padre morta improvvisamente a 58 anni) che poneva il forte sospetto di cardiomiopatia ipertrofica (HCM) sottostante. Inoltre, la paziente presentava in anamnesi l’intervento di tunnel carpale bilaterale e la rottura del capo-lungo del bicipite per traumatismo minore, entrambe red-flags per amiloidosi cardiaca in presenza di ipertrofia ventricolare sinistra (1).
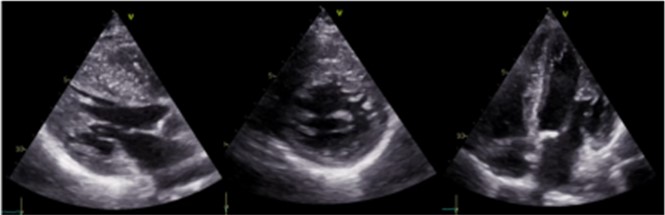
Inoltre, in considerazione della storia di SS con coinvolgimento multiorgano, una terza opzione possibile in grado di spiegare il quadro clinico della paziente si configurava nella cardiopatia sclerodermica a fenotipo ipertrofico.
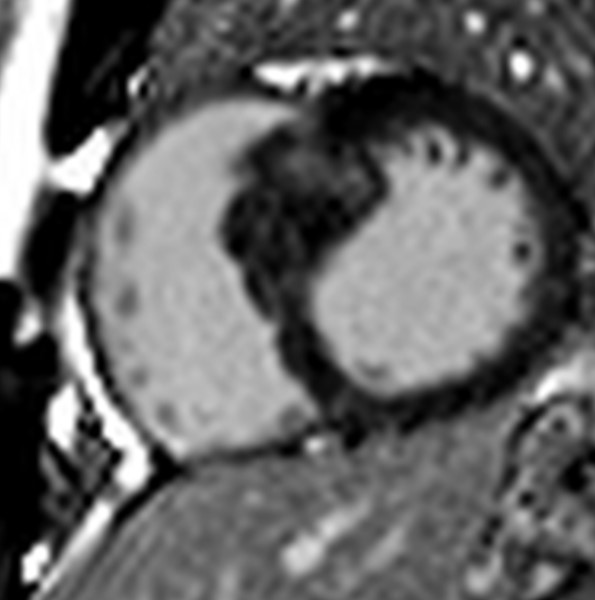
Per escludere l’opzione clinicamente più urgente, ossia l’amiloidosi cardiaca, venivano effettuate la scintigrafia miocardica con tracciante osseo e l’elettroforesi con immunofissazione sierica ed urinaria. Questi esami risultavano negativi, permettendoci così di escludere l’opzione dell’amiloidosi cardiaca. L’esame diagnostico successivo scelto fu la risonanza magnetica cardiovascolare, la quale, nonostante fosse limitata in termini di qualità dalla claustrofobia della paziente, confermava la marcata ipertrofia del ventricolo sinistro con prevalente coinvolgimento del setto interventricolare e repertava multiple aree di late gadolinium enhancement a distribuzione “patchy”, in assenza di edema (Figura 3).
Tali reperti di imaging, nonostante tipici della HCM (2), sono anche aspetto peculiare del coinvolgimento cardiaco da accumulo fibrotico della SS (3). Pertanto, per facilitare la diagnosi differenziale fra cardiopatia sclerodermica e coesistenza di HCM + SS, dopo aver ricostruito tre generazioni di albero genealogico senza individuare eventi chiave oltre a quello sopra-citato, la paziente veniva sottoposta ad un test genetico con panel customizzato per i geni responsabili delle cardiomiopatie strutturali.
In attesa dell’esito della genetica, veniva effettuata la stratificazione aritmica mediante l’esecuzione di un holter ECG delle 48 risultato negativo per eventi aritmici ventricolari ripetitivi. Sia lo Score multi-parametrico (HCM-SCD risk score) consigliato dalle Linee Guida Europee sull’HCM (4) che l’approccio “single major risk criteria” suggerito dalla Linee Guida Americane (5) rivelavano la non indicazione all’impianto di defibrillatore impiantabile in prevenzione primaria in caso di HCM. L’esito dell’ analisi genetica mostrava la presenza in eterozigosi della variante c.2167C> G nell’esone 20 del gene myosin heavy chain 7 (MYH7), mutazione responsabile di circa il 15-25% dei casi di HCM (6). Pertanto, l’esecuzione dell’analisi genetica ci ha permesso, non solo di effettuare una diagnosi complessa, ma anche di avviare lo screening fenotipico e genetico a cascata ai familiari di primo grado della paziente, screening tuttora in corso.
Discussione e Conclusioni
Nella pratica clinica ci si trova quotidianamente ad affrontare il dilemma fra singola malattia in grado di spiegare una presentazione clinica complessa e la coesistenza contemporanea di più malattie responsabili. Sono due gli approcci clinico-filosofici contrapposti. Il primo, inconsciamente ispirato al “Rasoio di Guglielmo di Occam” (7), il quale suggerisce che la spiegazione più semplice sia quella da preferire e che pertanto sia più probabile che una sola malattia spieghi in toto la clinica del paziente. Il secondo approccio invece, sulla scia della Legge di Murphy (8) (“se qualcosa può andare storto, lo farà”) suggerisce che una manifestazione clinica complessa sia più probabilmente da imputare alla coesistenza di più malattie sottostanti. Nel caso clinico della nostra paziente tre erano le diagnosi differenziali principali: cardiopatia sclerodermica a fenotipo ipertrofico, sclerosi sistemica associata ad amiloidosi cardiaca, sclerosi sistemica associata a cardiomiopatia ipertrofica. In tutti e tre i casi si trattava di entità cliniche relativamente rare. In Nord Italia la prevalenza di SS è di 1:400 soggetti, di cui circa il 30% mostrano un coinvolgimento cardiaco ( in primis l’ipertensione arteriosa polmonare), ossia 1: 1250 individui (9). Il fenotipo ipertrofico della cardiopatia sclerodermica è una condizione clinica solo riportata in letteratura, di cui non è nota la prevalenza e la cui peculiarità anatomo-patologica principale è l’accumulo a mosaico del materiale fibrotico nel miocardio (10) (11). Invece, l’associazione probabilistica fra SS e amiloidosi cardiaca è stimabile attorno ad 1:10’000’000 soggetti (1), mentre quella fra SS ed HCM attorno ad 1: 200 000 soggetti (5). Soltanto un approccio di imaging multimodale sequenziale combinato all’esecuzione dell’analisi genetica ci ha permesso di effettuare una diagnosi complessa e di iniziare lo screening genetico e fenotipico dei famigliari di primo grado. Pertanto, nel caso da noi riportato, la Legge di Murphy ha avuto la meglio su Rasoio di Occam.
Bibliografia
1. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42(16).
2. Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy: Part I, MRI appearances. Vol. 189, American Journal of Roentgenology. 2007.
3. Hachulla AL, Launay D, Gaxotte V, De Groote P, Lamblin N, Devos P, et al. Cardiac magnetic resonance imaging in systemic sclerosis: A cross-sectional observational study of 52 patients. Ann Rheum Dis. 2009;68(12).
4. Zamorano JL, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Vol. 35, European Heart Journal. 2014.
5. Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020.
6. Ingles J, Sarina T, Yeates L, Hunt L, Macciocca I, Mccormack L, et al. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15(12).
7. Wildner M. In memory of William of Occam [16]. Vol. 354, Lancet. 1999.
8. Scott JP, Ayano C, Sulman CG, Ruiz JP. Murphy’s law and Murphy eyes. Vol. 32, Journal of Anesthesia. 2018.
9. Lo Monaco A, Bruschi M, La Corte R, Volpinari S, Trotta F. Epidemiology of systemic sclerosis in a district of northern Italy. Clin Exp Rheumatol. 2011;29(2 SUPPL. 65).
10. Moyssakis I, Papadopoulos DP, Anastasiadis G, Vlachoyannopoulos P. Hypertrophic cardiomyopathy in systemic sclerosis. A report of two cases. Clin Rheumatol. 2006;25(3).
11. Sogomonian R, Alkhawam H, Lee S, Chang D, Moradoghli Haftevani E. ID: 6: HYPERTROPHIC OBSTRUCTIVE CARDIOMYOPATHY IN THE SETTING OF SYSTEMIC SCLERODERMA. J Investig Med. 2016;64(4).